2021-04-26-umich-rnaseqDemystified
MultiQC, Count Matrix, Wrap-Up
In this module, we will learn:
- how MultiQC gathers STAR alignment information, for QC purposes
- how MultiQC presents the results of STAR alignment
- how to combine gene-level results into a count matrix
Differential Expression Workflow
Here we will take the results from the previous module and operate on them a bit further. This will wrap up the day 1 exercises, leaving us well-poised to begin differential expression, which we will discuss on day 2.
| Step | Task |
|---|---|
| 1 | Experimental Design |
| 2 | Biological Samples / Library Preparation |
| 3 | Sequence Reads |
| 4 | Assess Quality of Raw Reads |
| 5 | Splice-aware Mapping to Genome |
| 6 | Count Reads Associated with Genes |
| :--: | ---- |
| 7 | Organize project files locally |
| 8 | Initialize DESeq2 and fit DESeq2 model |
| 9 | Assess expression variance within treatment groups |
| 10 | Specify pairwise comparisons and test for differential expression |
| 11 | Generate summary figures for comparisons |
| 12 | Annotate differential expression result tables |
Running MultiQC
After aligning reads it is often helpful to know how many reads were uniquely aligned, mapped to multiple loci, or not mapped at all. The sample_NLog.final.out file which is output alongside the alignments in sample_N.temp/ folder (we used the --keep-intermediate-files flag), reports this information:
Started job on | Oct 02 13:09:23
Started mapping on | Oct 02 13:09:51
Finished on | Oct 02 13:12:47
Mapping speed, Million of reads per hour | 238.82
Number of input reads | 11675504
Average input read length | 146
UNIQUE READS:
Uniquely mapped reads number | 10609591
Uniquely mapped reads % | 90.87%
Average mapped length | 146.37
Number of splices: Total | 2755543
Number of splices: Annotated (sjdb) | 2734730
Number of splices: GT/AG | 2739697
Number of splices: GC/AG | 13204
Number of splices: AT/AC | 1744
Number of splices: Non-canonical | 898
Mismatch rate per base, % | 0.18%
Deletion rate per base | 0.01%
Deletion average length | 1.60
Insertion rate per base | 0.01%
Insertion average length | 1.29
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 599915
% of reads mapped to multiple loci | 5.14%
Number of reads mapped to too many loci | 12786
% of reads mapped to too many loci | 0.11%
UNMAPPED READS:
Number of reads unmapped: too many mismatches | 20381
% of reads unmapped: too many mismatches | 0.17%
Number of reads unmapped: too short | 389960
% of reads unmapped: too short | 3.34%
Number of reads unmapped: other | 42871
% of reads unmapped: other | 0.37%
CHIMERIC READS:
Number of chimeric reads | 0
% of chimeric reads | 0.00%
We will run multiqc, and it will detect these reports from STAR and include them in the report.
MultiQC With STAR Exercise:
- Note the contents of our analysis directory, including the RSEM / STAR contents
- Run MultiQC on this directory
- Transfer the report back to local computer and view it
Click here for solution - MultiQC with STAR exercise
-
Note contents of our analysis directory, including RSEM / STAR contents
ls -l ~/analysis/rsem_star/ -
Run MultiQC on this directory
multiqc --outdir ~/analysis/multiqc_star ~/analysis/rsem_star -
Transfer the report back to local computer and view it
exit # log out from remote # Now on local scp <username>@50.17.210.255:~/analysis/multiqc_star/multiqc_report.html ~/workshop_rsd/multiqc_report_star.html
Use GUI file manager to find your ~/workshop_rsd folder. Double-click multiqc_report.html (open it with an internet browser).
The newly included STAR section will look something like the following:
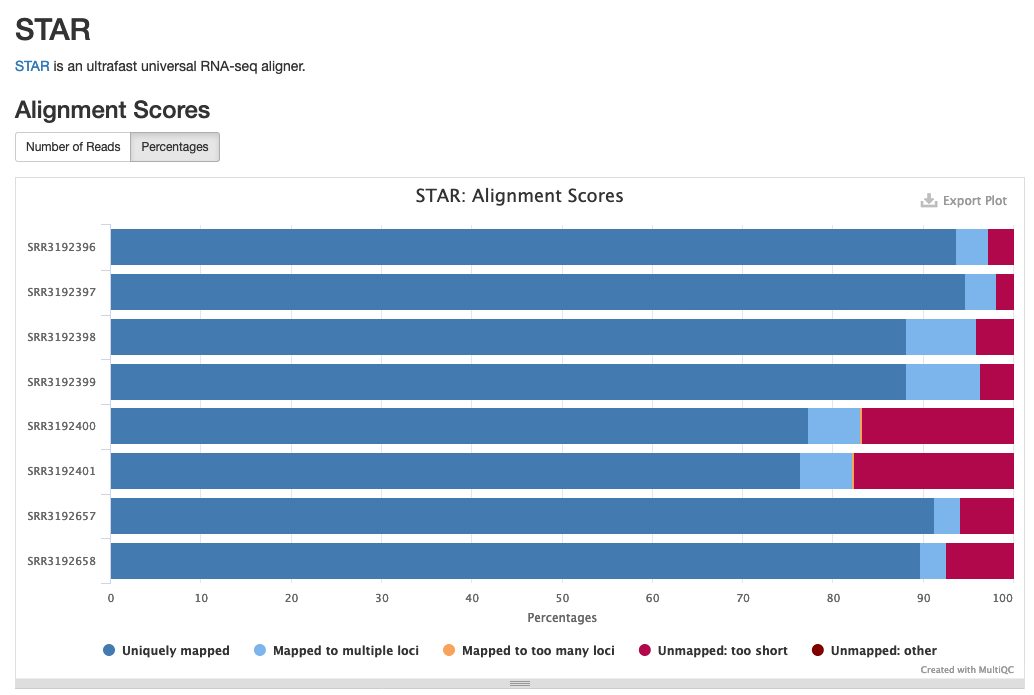 Example of STAR alignment statistics in MultiQC.
Example of STAR alignment statistics in MultiQC.
Source: MultiQC example report
Creating the count matrix
We have viewed some of the gene expression quantification results individually. It can be useful to combine these expression values into a count matrix. This is helpful when gathering expression-level QC metrics, as well as for input into a differential gene expression program such as DESeq2.
Count Matrix Exercise:
- View the
.genes.resultsfiles that we want to combine - Understand the process of creating a count matrix
- View the help file of
combine.py - Construct / execute a command to combine our results into a count matrix
- View the resulting count matrix
Click here for solution - Creating count matrix exercise
-
Log back in to aws instance with
ssh <username>@50.17.210.255 -
View the
.genes.resultsfiles that we want to combinehead -n 1 ~/analysis/rsem_star/sample_01.genes.results # It's easiest to look at the first line (header) -
Understand the process of creating a count matrix
-
View the help file of
combine.pycombine.py --help -
Construct / execute a command to combine our results into a count matrix
combine.py --output_file ~/analysis/count_matrix.tsv --input_path 'analysis/rsem_star/*.genes.results' --column expected_count --id_columns gene_id -
View the resulting count matrix
head ~/analysis/count_matrix.tsv
Contents of combine.py script
There are many ways to combine these results into a count matrix. Here is how this python script we've used, `combine.py`, works:
'''Combines the count/FPKM/TPM from individual sample outputs into one matrix'''
import argparse
from glob import glob
from os.path import commonprefix, basename
import re
import sys
import numpy as np
import pandas as pd
__version__ = '0.0.1'
_DESCRIPTION = \
'''Accepts tab-separated sample isoform files and combines into single tab-separated matrix.'''
def _commonsuffix(strings):
return commonprefix(list(map(lambda s:s[::-1], strings)))[::-1]
def _build_sample_files(file_glob):
sample_files = []
file_names = glob(file_glob)
suffix = _commonsuffix(file_names)
for file_name in sorted(file_names):
sample_name = basename(file_name).replace(suffix, '')
sample_files.append((sample_name, file_name))
return sample_files
def _parse_command_line_args(sys_argv):
parser = argparse.ArgumentParser(
description=_DESCRIPTION)
parser.add_argument(
'-o', '--output_file',
type=str,
help='path to combined output file',
required=True)
parser.add_argument(
'-i', '--input_path',
type=str,
help='path (including linux wildcards) to sample input files; surround with single quotes when usimg wildcards',
required=True)
parser.add_argument(
'-c', '--column',
type=str,
help='full name of column to extract from inputs (e.g. FPKM)',
required=True)
parser.add_argument(
'--id_columns',
type=str,
help='gene_id or gene_id,transcript_id',
required=True)
parser.add_argument('--version',
'-V',
action='version',
version=__version__)
args = parser.parse_args(sys_argv)
args.id_columns=args.id_columns.split(',')
return args
def main(argv):
print('combine v{}'.format(__version__))
print('command line args: {}'.format(' '.join(argv)))
args = _parse_command_line_args(argv[1:])
sample_files = _build_sample_files(args.input_path)
output_filename = args.output_file
merge_column = args.column
name, file = sample_files.pop(0)
df=pd.read_csv(file, sep='\t', low_memory=False)
# Round expected counts and convert to integers
df['expected_count'] = np.rint(df['expected_count']).astype(int)
new=pd.DataFrame(df[args.id_columns+[merge_column]])
new.rename(columns={merge_column:name},inplace=True)
for (name, file) in sample_files:
df=pd.read_csv(file, sep='\t')
df['expected_count'] = np.rint(df['expected_count']).astype(int)
new[name]=df[merge_column]
print('saving {} ({} x {})'.format(output_filename, *new.shape))
new.to_csv(output_filename,sep='\t',index=False)
print('done')
if __name__ == '__main__':
main(sys.argv)
These materials have been adapted and extended from materials created by the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.