Module 07: DE Initialization
UM Bioinformatics Core
2023-03-14
Objectives
- ‘Unblind’ our sample IDs
- Understand possible confounding factors
- Understand the impact of batches or additional covariates
- Filter count table
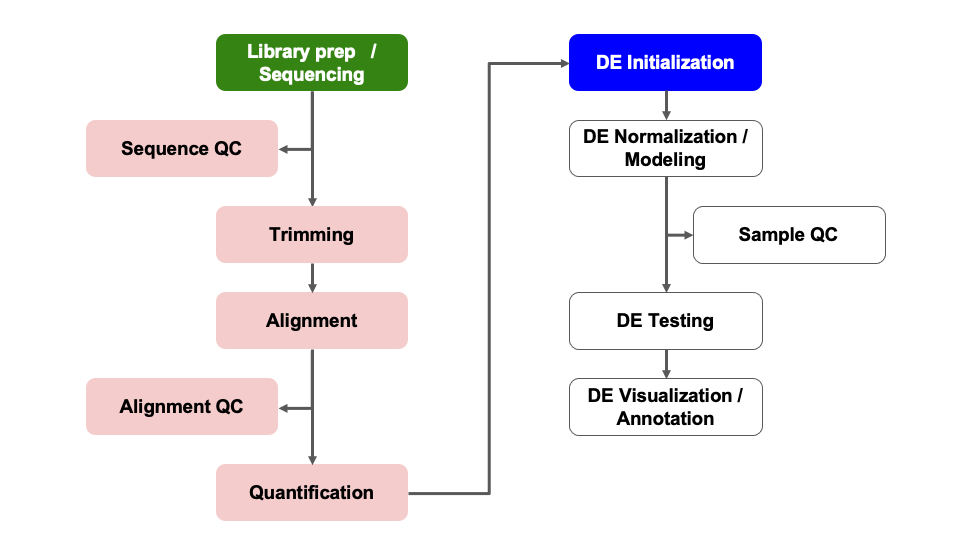
Differential Expression Workflow
Here we will proceed setting up the inputs needed to initialize DESeq2 before testing for differential expression.
DESeq2 objects
Bioconductor software packages often define and use custom classes within R to store data in a way that better fits expectations around biological data, such as illustrated below from Huber et al. 2015.
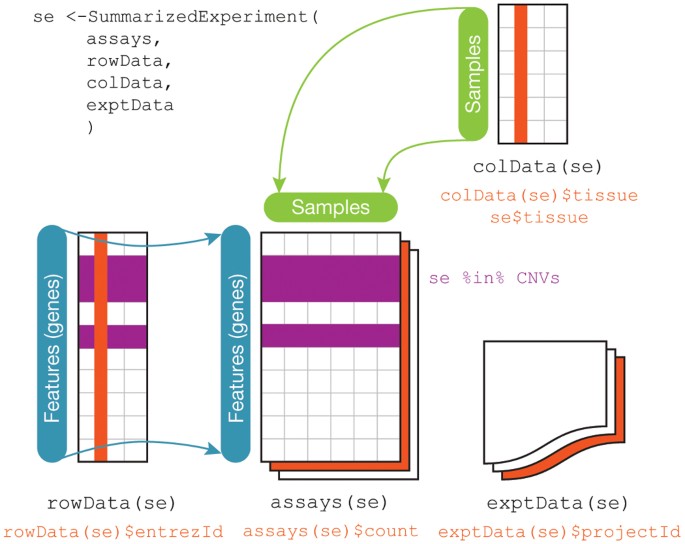
A breakdown of the SummarizedExperiment class.
These custom data structures have pre-specified data slots, which hold specific types/classes of data and therefore can be more easily accessed by functions from the same package.
To create the DESeqDataSet we need two tables:
- A count matrix (which we already loaded and saved as the object
count_table) and - A table that assigns the condition labels for each sample (which we will generate below).
Sample Information
As introduced at the beginning of the workshop, we have downloaded and prepared data from an existing publication (Zhang et al., 2019), wherein one goal is to understand the gene expression differences in wild-type mice that are “iron replete” (we label these “plus”) and “iron deficient” (we label these “minus”).
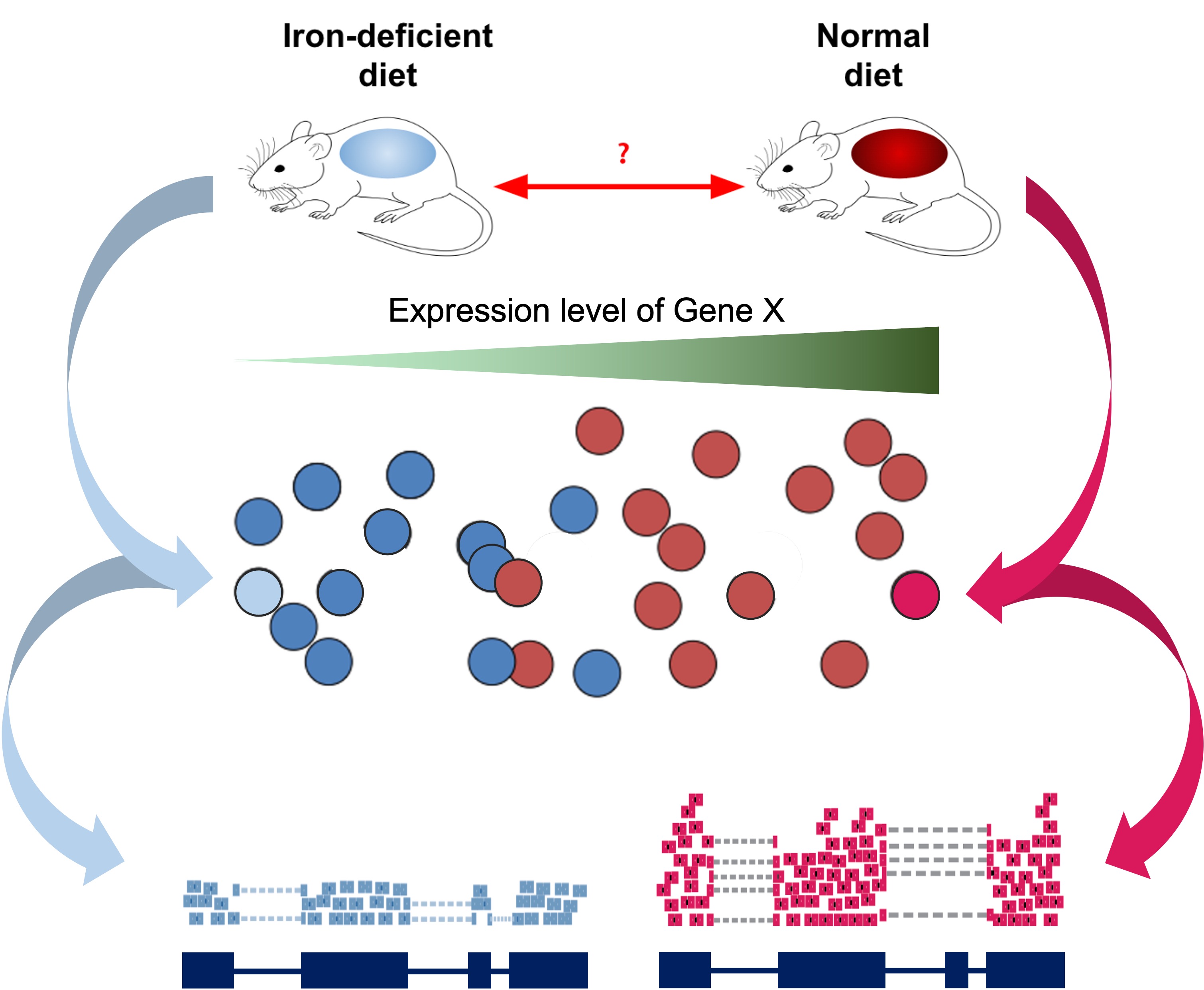
Read Sample Table
Our next step will be to describe the samples within our R session, so that we make the proper comparisons with DESeq2. Let’s check the sample names from the count table.
colnames(counts_rounded)[1] "sample_A" "sample_B" "sample_C" "sample_D" "sample_E" "sample_F"Based on the sample names that are the columns of
counts_rounded, our samples are blinded, e.g. the sample
names don’t clearly correspond to treatment groups. We will need to
specify which sample IDs connect to which experimental conditions.
Typically sample phenotype data, including experimental conditions, are stored as Excel or CSV files that we can read into R, and then use when creating a DESeq2 object. If you are unfamilar with CSV files or how to generate them, there are tutorials available to guide you through the process.
Tip: Sample naming conventions
Use only alpha-numeric characters (A-Z, a-z, 0-9), and separate parts of the name with underscores (
_) or dots (.). Do not begin sample names with numbers.
We’ll load our ‘pre-made’ sample information sheet,
samplesheet.csv, to unblind our samples.
samplesheet = read.table("data/samplesheet.csv",
sep = ",",
header = TRUE,
row.names = 1)We can look at the object by typing its name and hitting Enter. Note,
for larger experiments, you may want to use head().
samplesheet genotype condition
sample_A wt plus
sample_B wt plus
sample_C wt plus
sample_D wt minus
sample_E wt minus
sample_F wt minusCheckpoint: If you have loaded
samplesheet, please indicate with the green ‘check’ button.
Otherwise, please use the red ‘x’ button to have the command
repeated
In this example data, all mice are wild-type and the iron replete
mice are labeled as “plus” and the iron deficient mice are labeled as
“minus”. Again, for larger experiments, you may want to examine the
coding of the samples by looking at the unique() values of
the relevant columns.
unique(samplesheet$condition)[1] "plus" "minus"Replicates in RNA-seq experiments
A frequent question for RNA-seq projects is “How many replicates do I need?”.
The general goal of our analyses is to separate the “interesting” biological contributions from the “uninteresting” technical or other contributions that either cannot be or were not controlled in the experimental design. The more sources of variation, such as samples coming from heterogenous tissues or experiments with incomplete knockdowns, the more replicates (>3) are recommended.
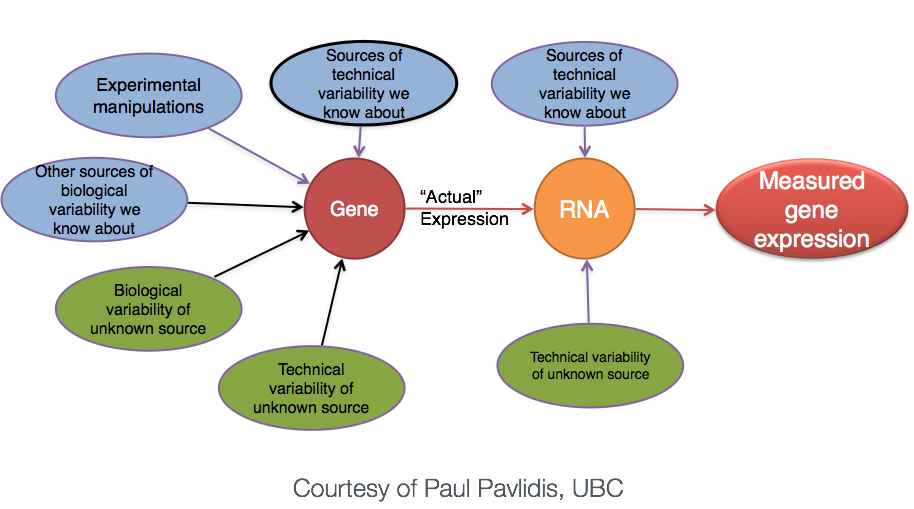
Image of technical, biological, and experimental contributors to gene expression, from HBC training materials
For a more in depth discussion of experimental design considerations, particularly for the number of replicates, please read A Beginner’s Guide to Analysis of RNA Sequencing Data and papers like this one by Hart et al that focus on estimating statistical power for RNA-seq experiments.
Sequencing depth recommendations
A related experimental design consideration we often discuss with researchers is how much sequencing depth should be generated per sample. This figure shared by Illumina in their technical talks is helpful to understand the relative importance of replicates versus sequencing depth.
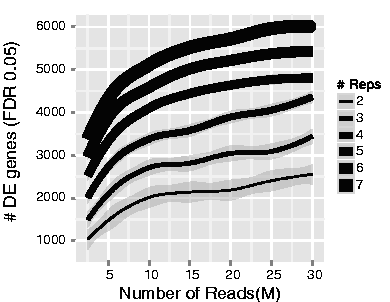
Illumina’s differential expression recovery across replicate number and sequencing depth
Generally, for human and mouse experiments the recommendation is 30-40 million reads per sample to capture both highly expressed (abundant) and lowly expressed (rarer) transcripts, assuming that ~25,000 protein-coding genes would be measured for a polyA library prep. However, as the image above shows, sequencing depth has less of an impact than number of replicates in detecting differentially expressed genes (DEGs).
Sample table formatting
Next, we’ll format our table so that we have the appropriate data type (an ordered factor) for DESeq2 to recognize our treatment groups and appropriately compare samples.
# tidy version
samplesheet_ready <- samplesheet %>% mutate(condition = factor(condition,
levels = c('plus', 'minus')))
# base version
samplesheet_ready <- samplesheet
samplesheet_ready$condition = factor(samplesheet_ready$condition,
levels = c('plus', 'minus'))
unique(samplesheet_ready$condition)[1] plus minus
Levels: plus minusNotice that we set the levels in a particular order. This is important for setting the “Control” (or “Reference”) group as the denominator in the default comparisons when we setup our DESeq2 model.
Before we proceed, we need to make sure that the sample labels (column names) in the count table match the sample information table (row names), including the order. If the sample labels don’t match, then we will see an error and need to correct the labels prior to proceeding. Checking the sample information table is extremely important to ensure that the correct samples are grouped together for comparisons.
all(colnames(count_table) == rownames(samplesheet_ready))[1] TRUEThis line of code checks if both the identity and order match between
our count_table and our samplesheet. If, in
the course of using your own data, this returns FALSE, try
using the match() function to rearrange the columns of
count_table (or the rows of samplesheet) to
get them to match.
Checkpoint: If you your sample info check
returns TRUE, please indicate with the green ‘yes’ button.
Otherwise, please use the red ‘x’ button to have the command
repeated
Creating DESeq2 object
To create the DESeqDataSet we will need the
count_table and the samplesheet. We will also
need a design formula to specify our model.
Making model choices
The design formula specified informs many of the DESeq2 functions how to treat the samples in the analysis, specifically which column(s) in the sample metadata table are relevant to the experimental design.
In this case, we aren’t aware of any covariates that should be considered in our comparisons. However, if there are additional attributes of the samples that may impact the DE comparisons, like date of collection, sex, or patient of origin, these should be added as additional columns in the sample information table and added to a design formula.
More complex questions, including adding “interaction terms” between multiple group labels are helpfully described in this support thread as well as in the DESeq2 vignette, which is an excellent document.
The design formula specifies the column(s) in the metadata table and
how they should be used in the analysis. For our dataset we only have
one column we are interested in, that is condition. This
column has two factor levels, which tells DESeq2 that for each gene we
want to evaluate gene expression change with respect to these different
levels.
## Create DESeq object, line by line
dds = DESeqDataSetFromMatrix(countData = counts_rounded,
colData = samplesheet_ready,
design = ~ condition)converting counts to integer modeddsclass: DESeqDataSet
dim: 55492 6
metadata(1): version
assays(1): counts
rownames(55492): ENSMUSG00000000001 ENSMUSG00000000003 ... mcherry tdtomato
rowData names(0):
colnames(6): sample_A sample_B ... sample_E sample_F
colData names(2): genotype conditionNotice that printing the dds object helpfully shows us
some helpful information:
- The dimension (number of genes by number of samples),
- The gene identifiers,
- The sample identifiers,
- The additional column names giving information about the samples
Checkpoint: If you see dds in your
environment panel, please indicate with the green ‘check’ button.
Otherwise, please use use the red ‘x’ button in your zoom reaction panel
to have this step repeated. You can use the red ‘x’ to be put in a
breakout room for help
Pre-filtering
While not necessary, pre-filtering the count table helps to not only reduce the size of the DESeq2 object, but also gives you a sense of how many genes were reasonably measured at the sequencing depth generated for your samples.
Here we will filter out any genes that have less than 10 counts across any of the samples. This is a fairly standard level of filtering, but can filter data less/more depending on quality control metrics from alignments and sequencing depth or total number of samples.
keep = rowSums(counts(dds)) >= 10
dds_filtered = dds[keep,]
dds_filteredclass: DESeqDataSet
dim: 16249 6
metadata(1): version
assays(1): counts
rownames(16249): ENSMUSG00000000001 ENSMUSG00000000028 ... ENSMUSG00000118651
ENSMUSG00000118653
rowData names(0):
colnames(6): sample_A sample_B ... sample_E sample_F
colData names(2): genotype conditionNotice the dds_filtered object has less elements than
the unfiltered dds object, indicating that a number of
genes were not measured in our experiment.
Checkpoint: Questions?
Optional content
Click for how to model batch effects with DESeq2
Differences between samples can also be due to technical reasons, such as collection on different days or different sequencing runs. Differences between samples that are not due to biological factors as called batch effects. We can include batch effects in our design model in the same way as covariates, as long as the technical groups do not overlap, or confound, the biological treatment groups.
Let’s try add some additional meta-data information where we have counfounding batch effects and create another DESeq2 object.
samplesheet_batch = samplesheet_ready
samplesheet_batch$batch = factor(c(rep(c("Day1"), 2),
rep(c("Day2"), 2),
rep(c("Day3"), 2)),
levels = c("Day1", "Day2", "Day3"))
dds_batch = DESeqDataSetFromMatrix(countData = counts_rounded,
colData = samplesheet_batch,
design = ~ batch + condition)converting counts to integer modeSummary
In this section, we:
- Loaded the necessary input files into our R session
- Discussed model design for DESeq2
- Initialized a DESeq2 data set
- Filtered our count data
Now that we’ve created our DESeq2 objects, including specifying what model is appropriate for our data, and filtered our data, we can proceed with assessing the impact of the experimental conditions on gene expression for our samples.
Sources
Training resources used to develop materials:
- HBC DGE setup: https://hbctraining.github.io/DGE_workshop/lessons/01_DGE_setup_and_overview.html
- HBC Count Normalization: https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
- DESeq2 standard vignette: http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
- DESeq2 beginners vignette: https://bioc.ism.ac.jp/packages/2.14/bioc/vignettes/DESeq2/inst/doc/beginner.pdf
- Bioconductor RNA-seq Workflows: https://www.bioconductor.org/help/course-materials/2015/LearnBioconductorFeb2015/B02.1_RNASeq.html
- CCDL Gastric cancer training materials: https://alexslemonade.github.io/training-modules/RNA-seq/03-gastric_cancer_exploratory.nb.html
- CCDL Neuroblastoma training materials: https://alexslemonade.github.io/training-modules/RNA-seq/05-nb_cell_line_DESeq2.nb.html
These materials have been adapted and extended from materials listed above. These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.