DE Normalization
UM Bioinformatics Core
2024-07-31
In this module, we will learn:
- Different count normalizations and their uses
- How to fit a model for differential expression comparisons
Differential Expression Workflow
Here we will proceed with count normalizations.
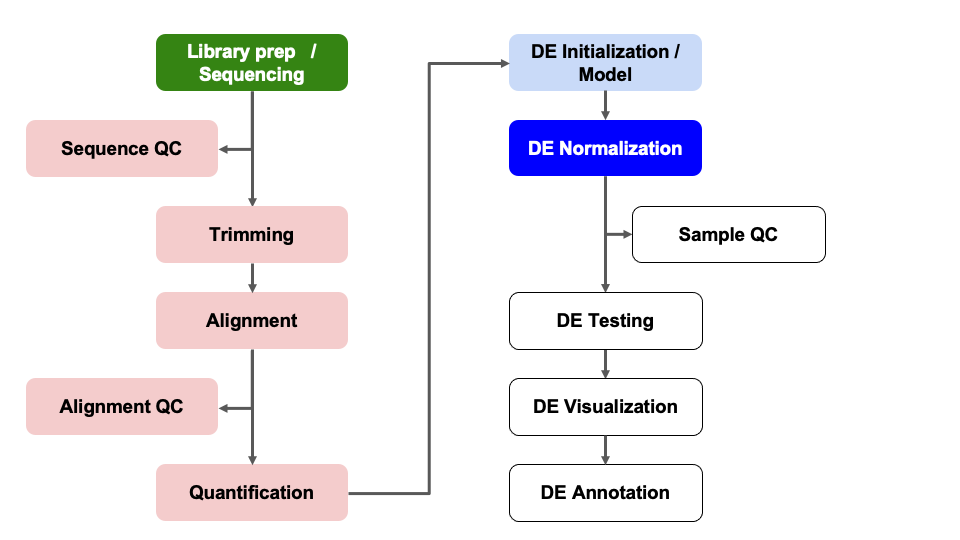
Count normalizations
Since counts of mapped reads for each gene is proportional to the expression of RNA in addition to many “uninteresting” other factors, normalization is the process of scaling raw count values to account for the “uninteresting” factors and ensure expression levels are more comparable.
Normalization goals
Two common factors that need to be accounted for during normalization are sequencing depth and gene length. Common normalization approaches (such as FPKM, RPKM, CPM, TPM, etc.) account for one or both of these factors.
- Sequencing depth normalization is necessary to account for the proportion of reads per gene expected for more deeply sequenced samples (like in pink below) versus a less deeply sequenced sample (like in green below).
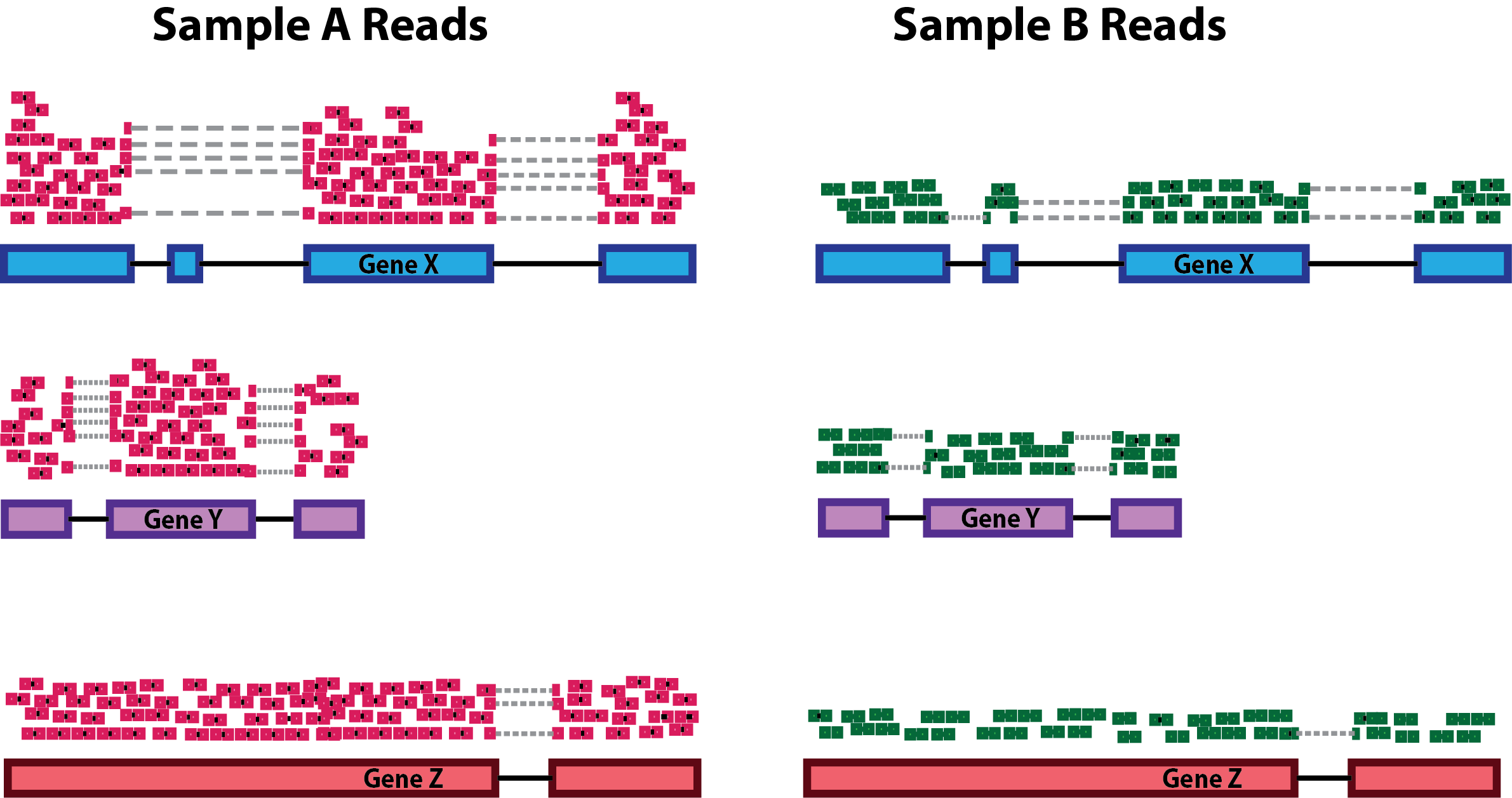
- Gene length normalization may also be necessary if comparing between different genes, since genes of different lengths have different probabilities of generating fragments that end up in the library.
Note: The above figure was originally from a HBC tutorial that also includes a detailed comparison of different normalization (CPM, TPM, FPKM) approaches and their best uses, which are summarized below.
Common normalization methods
Several common normalization methods exist to account for these differences:
| Normalization method | Description | Accounted factors | Recommendations for use |
|---|---|---|---|
| CPM (counts per million) | counts scaled by total number of reads | sequencing depth | gene count comparisons between replicates of the same samplegroup; NOT for within sample comparisons or DE analysis |
| TPM (transcripts per kilobase million) | counts per length of transcript (kb) per million reads mapped | sequencing depth and gene length | gene count comparisons within a sample or between samples of the same sample group; NOT for DE analysis |
| RPKM/FPKM (reads/fragments per kilobase of exon per million reads/fragments mapped) | similar to TPM | sequencing depth and gene length | gene count comparisons between genes within a sample; NOT for between sample comparisons or DE analysis |
| DESeq2’s median of ratios [1] | counts divided by sample-specific size factors determined by median ratio of gene counts relative to geometric mean per gene | sequencing depth and RNA composition | gene count comparisons between samples and for DE analysis; NOT for within sample comparisons |
| EdgeR’s trimmed mean of M values (TMM) [2] | uses a weighted trimmed mean of the log expression ratios between samples | sequencing depth, RNA composition | gene count comparisons between samples and for DE analysis; NOT for within sample comparisons |
Check-in: Questions about normalizations?
DESeq2 normalizations
DESeq2 has an internal normalization process that accounts for RNA composition. A few highly differentially expressed genes, differences in the number of genes expressed between samples, or contamination are not accounted for by depth or gene length normalization methods. Accounting for RNA composition is particularly important for differential expression analyses, regardless of the tool used.
For data exploration and visualizations, it is helpful to generate an object of independently normalized counts. We will use the rlog transformation from DESeq2 that accounts for sequencing depth for each sample and RNA composition for the downstream quality control visualizations.
The rlog transformation produces log2 scaled data that has also been normalized to overall library size as well as variance across genes at different mean expression levels. For larger numbers of samples, there is an alternative transformation method, vst that can be used instead for count normalization.
The command to generate the normalized count object has a few parts,
including dds as an input and providing a value to the
option blind and then we’ll look at the results of the
transformation by extracting the values with the assay()
function.
# create object with rlog normalized data
rld = rlog(dds_filtered, blind = TRUE)
# check contents of new object `rld`
head(assay(rld), 2) sample_A sample_B sample_C sample_D sample_E sample_F
ENSMUSG00000000001 10.51481 10.36671 10.41946 10.84037 10.41045 10.57877
ENSMUSG00000000028 10.60446 10.73451 10.73503 10.68271 10.82094 10.99100Looking at the rld values, we can see that they are now in log scale.
Since we set blind=TRUE, the transformation is blind to the
sample information we specified in the design formula. The normalized
counts are helpful for visualization methods during expression-level
quality assessment but aren’t used in the model
fitting.
We’ll come back to these normalized data, but first let’s write out both the raw and normalized count tables to file.
Output count tables
To output the raw counts, we will need to use the counts
function to access the count table from within its larger
DESeqDataSet object. Then we’ll output the rlog count
table, using the assay function to access the normalized
count table from within its larger DESeqDataSet object.
# output raw counts to file
write.csv(counts(dds, normalized = FALSE), file="outputs/tables/DESeq2_raw_counts.csv")
# output rlog normalized counts to file
write.csv(assay(rld), file="outputs/tables/DESeq2_rlog_normalized_counts.csv")Summary
In this section, we:
- Learned about count normalizations and uses
- Generated a normalized count table
- Wrote intermediate data to file
Next, we’ll generate sample level quality control visualizations.
Sources
Training resources used to develop materials
- HBC DGE setup: https://hbctraining.github.io/DGE_workshop/lessons/01_DGE_setup_and_overview.html
- HBC Count Normalization: https://hbctraining.github.io/DGE_workshop/lessons/02_DGE_count_normalization.html
- DESeq2 standard vignette: http://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
- DESeq2 beginners vignette: https://bioc.ism.ac.jp/packages/2.14/bioc/vignettes/DESeq2/inst/doc/beginner.pdf
- Bioconductor RNA-seq Workflows: https://www.bioconductor.org/help/course-materials/2015/LearnBioconductorFeb2015/B02.1_RNASeq.html
These materials have been adapted and extended from materials listed above. These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
| Previous lesson | Top of this lesson | Next lesson |
|---|