RNA-seq Demystified Workshop
UM Bioinformatics Core
Wrapping up
We hope you now have more familiarity with key concepts, data types, tools, and how they all connect to enable gene expression analysis from bulk RNA-Seq data.
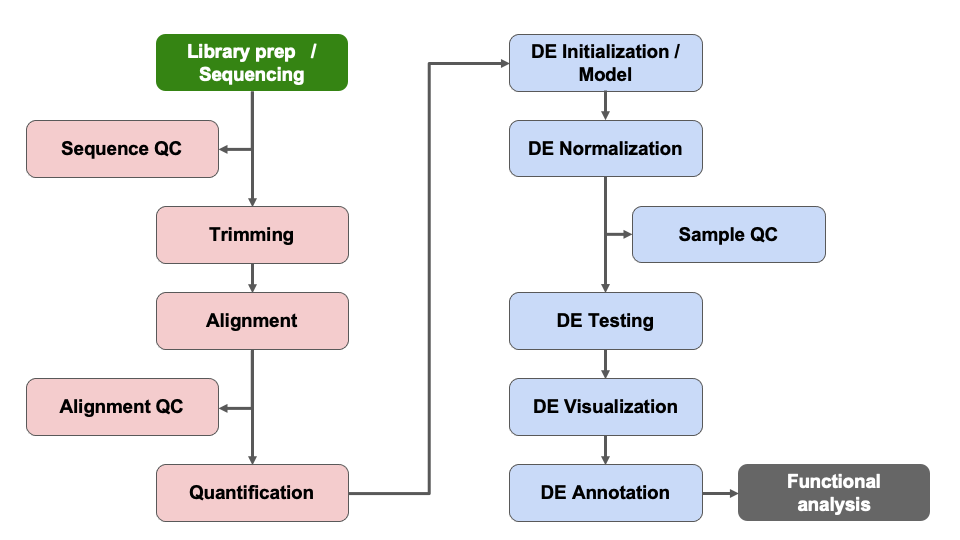
A method-style summary & R session info
The reads were trimmed using Cutadapt v2.3 (Martin, 2011). FastQC v0.11.8 was used to ensure the quality of data (Andrews, 2010). Reads were mapped to the reference genome GRChm38 (ENSEMBL Release 102), using STAR v2.7.8a (Dobin et al., 2013) and assigned count estimates to genes with RSEM v1.3.3 (Li and Dewey, 2011). Alignment options followed ENCODE standards for RNA-seq. Multiqc v1.7 compiled the results from several of these tools and provided a detailed and comprehensive quality control report (Ewels et al., 2016).
Data were pre-filtered to remove genes with 10 counts in all samples. Differential gene expression analysis was performed using DESeq2 (Love et al., 2014), using a negative binomial generalized linear model (thresholds: linear fold change >1.5 or <-1.5, Benjamini-Hochberg FDR (Padj) <0.05). Plots were generated using variations of DESeq2 plotting functions and other packages with R version 4.4.1. Annotation data from GRCm38 (Ensembl release 102) was used, and genes were additionally annotated with Entrez GeneIDs and text descriptions.
Session Info
------------
R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 20.04.6 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8 LC_COLLATE=C.UTF-8
[5] LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8 LC_PAPER=C.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: America/Detroit
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] plotly_4.10.4 data.table_1.15.4 RColorBrewer_1.1-3
[4] pheatmap_1.0.12 ggrepel_0.9.5 lubridate_1.9.3
[7] forcats_1.0.0 stringr_1.5.1 dplyr_1.1.4
[10] purrr_1.0.2 readr_2.1.5 tidyr_1.3.1
[13] tibble_3.2.1 ggplot2_3.5.1 tidyverse_2.0.0
[16] DESeq2_1.44.0 SummarizedExperiment_1.34.0 Biobase_2.64.0
[19] MatrixGenerics_1.16.0 matrixStats_1.3.0 GenomicRanges_1.56.1
[22] GenomeInfoDb_1.40.1 IRanges_2.38.1 S4Vectors_0.42.1
[25] BiocGenerics_0.50.0
loaded via a namespace (and not attached):
[1] gtable_0.3.5 htmlwidgets_1.6.4 lattice_0.22-6
[4] tzdb_0.4.0 vctrs_0.6.5 tools_4.4.1
[7] generics_0.1.3 parallel_4.4.1 fansi_1.0.6
[10] pkgconfig_2.0.3 Matrix_1.7-0 lifecycle_1.0.4
[13] GenomeInfoDbData_1.2.12 compiler_4.4.1 munsell_0.5.1
[16] codetools_0.2-20 htmltools_0.5.8.1 lazyeval_0.2.2
[19] pillar_1.9.0 crayon_1.5.3 BiocParallel_1.38.0
[22] DelayedArray_0.30.1 abind_1.4-5 digest_0.6.36
[25] tidyselect_1.2.1 locfit_1.5-9.10 stringi_1.8.4
[28] fastmap_1.2.0 grid_4.4.1 colorspace_2.1-0
[31] cli_3.6.3 SparseArray_1.4.8 magrittr_2.0.3
[34] S4Arrays_1.4.1 utf8_1.2.4 withr_3.0.0
[37] scales_1.3.0 UCSC.utils_1.0.0 timechange_0.3.0
[40] XVector_0.44.0 httr_1.4.7 hms_1.1.3
[43] viridisLite_0.4.2 rlang_1.1.4 Rcpp_1.0.13
[46] glue_1.7.0 rstudioapi_0.16.0 jsonlite_1.8.8
[49] R6_2.5.1 zlibbioc_1.50.0 Housekeeping
Please take our optional post-workshop survey (5-10 minutes)
We will email you a link to the final session recordings next week.
The website/notes for this workshop will be available.
The UM Bioinformatics Core Workshop Slack channel will be available for 90 days.
Looking ahead
Workshop environment
BASH/RStudio workshop compute environment will be available until 8/31.
- Please save all your R scripts now so that we can “right-size” the compute environment immediately following today’s workshop session.
You can download files from the workshop environment from your terminal/command line window as below. (You will need to substitute your actual workshop username and type workshop password when prompted.)
mkdir rnaseq-demystified-workshop cd rnaseq-demystified-workshop scp -r YOUR_USERNAME@bfx-workshop01.med.umich.edu:"RSD*" .- Note that the full download is about 2-4Gb, so depending on your internet speeds it could take while.
Installing software locally
- You can install necessary programs to run programs locally. Note
that for typical data, the compute intensive steps (Day 1) assume your
computer has powerful compute (many CPUs and lots of RAM) and sizable
storage capacity. (i.e. It may not be practical to run these on your
laptop.) Also, Installing bioinformatics software is non-trivial and
comprehensive instructions to setup a complete compute environment are
outside the scope of this workshop. For the intrepid:
- For UM folks interested in learning more about the GreatLakes HPC and more advanced software installation approaches, see our workshop lessons on Reproducible Computing
- Instructions on Installing
Conda. And the command to create a similar Conda environment:
conda create -n rsd -c conda-forge -c bioconda multiqc fastqc cutadapt star rsem numpy pandas - Instructions on setting up R/R-Studio
University of Michigan Resources
- UM CoderSpaces “office hours” and UM CoderSpaces Slack workspace. (See “Useful Resources” section in CoderSpaces page for instructions on how to join and access the CoderSpaces Slack workspace.)
- Upcoming UM Advanced Research Computing workshops.
- Advanced Research Computing (ARC) at University of Michigan hosts a
high-performance computing (HPC) platform called Great Lakes
which combines high-end computers, fast/resilient storage, and
pre-installed software. GreatLakes may be a good resource for folks who
need to run the more compute intensive steps and a substantial block of
compute and storage is subsidized by ARC making it essentially free to
many researchers.
- About Great Lakes HPC.
- About the ARC Research Computing Package.
- Videos on getting started with Great Lakes. (available to UM folks)
Training and support
- Learning bioinformatic analyses more like a process than a task.
Resources to consider:
- UM Bioinformatics Core links to training resources/workshops
- Notes on Bash and R from the recent
Computational
Foundations Workshop.
- For more intro lessons and workshops in Bash / Git / R / Python : Software Carpentry and the UM Software Carpentry Group.
- For more info on NGS analysis, we can highly recommend training materials from the Harvard Chan Bioinformatics Core.
Thank you

Thank you to/from the workshop team
 |
 |
 |
 |
|---|---|---|---|
| Chris | Marci | Travis | Raymond |
 |
 |
 |
|
| Dana | Matt | Nick |
Thank you for participating in our workshop. We welcome your questions and feedback now and in the future.
Bioinformatics Workshop Team