Bulk RNA-Seq
UM Bioinformatics Core
2023-08-29
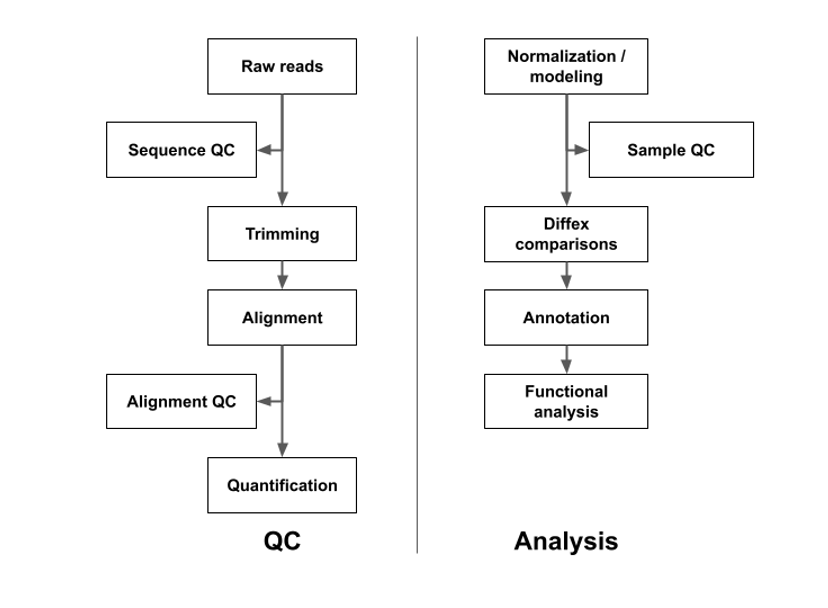
Methods
Adapters will be trimmed with CutAdapt (Martin) to remove adapters and low-quality bases and overall quality will assessed with FastQC (FastQC), FastQCScreen (Wingett and Andrews, 2018), and MultiQC (Ewels et al., 2016). Reads will be aligned and quantified using RSEM/STAR (Li and Dewey, 2011; Dobin et al., 2013). Differential expression modeling will use the DESeq2 (Love et al., 2014). Intra and inter group variance will be assessed with Principal Component Analysis. Candidate DEGs are visualized with volcano plots filtered to the subset of DEGs where Benjamini-Hochberg adjusted p-value ≤ 0.05 (Benjamini and Hochberg, 1995). Functional analysis using Advaita iPathwayGuide (Draghici et al., 2007; iPathwayGuide) will assess enrichment of KEGG pathways and Gene Ontology (GO) concepts.