Bulk DNA Methylation
UM Bioinformatics Core
2023-08-29
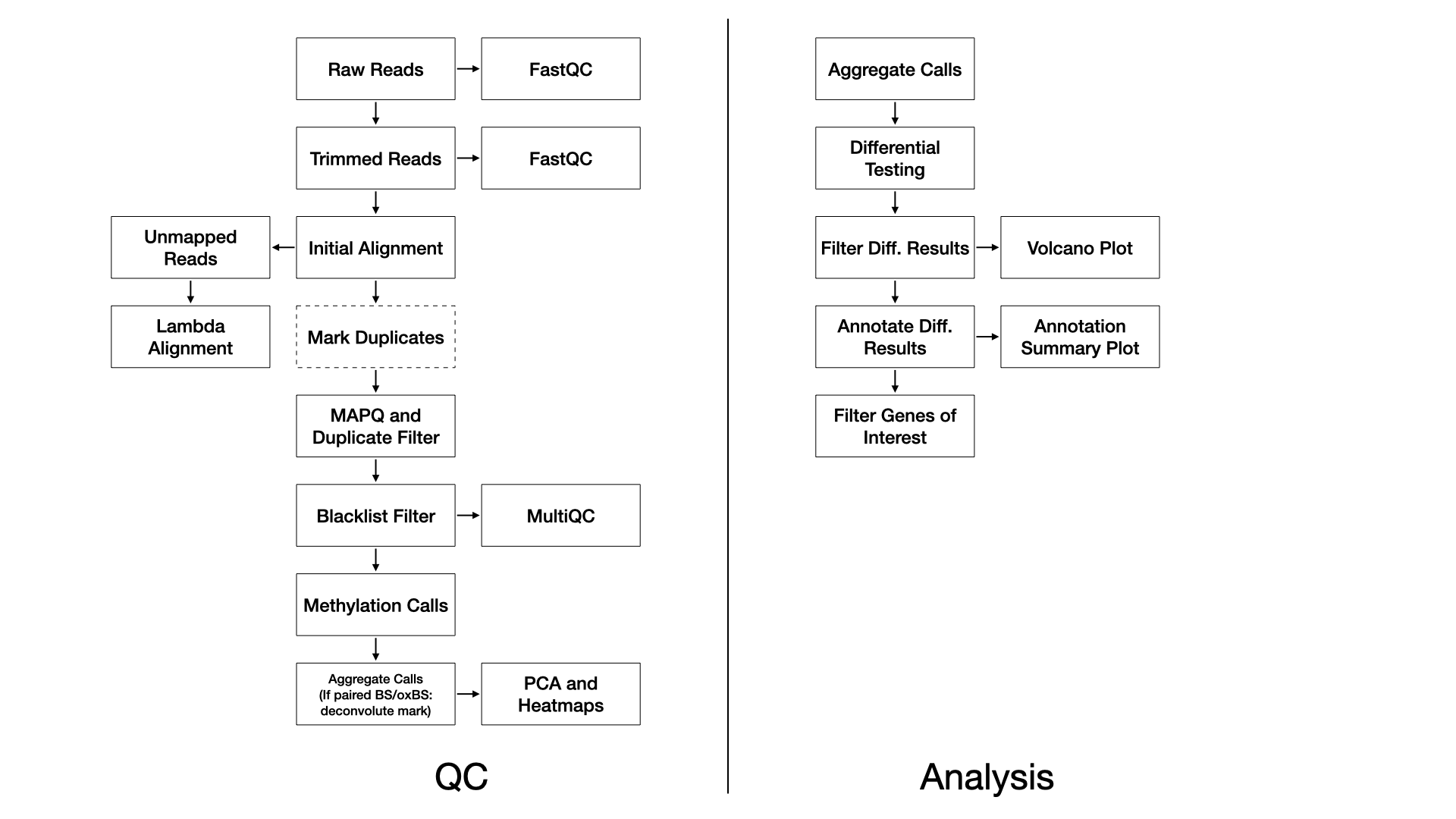
Methods
QC
We use FastQC (FastQC) (v0.11.8) to assess the overall quality of each sequenced sample.
We use TrimGalore (@
article?{noauthor_trimgalore_nodate,) (v0.4.5)
with the following parameters:
--adapter AGATCGGAAGAGC -e 0.1 --stringency 6 --length 20 --nextseq 20,
and with the additional --rrbs flag in the ERRBS case.
Reads are aligned to the reference genome with Bismark (Krueger and Andrews, 2011) (v0.22.1) using Bowtie2 (Langmead and Salzberg, 2012) (v2.3.4) with default settings (multi-seed length of 20bp with 0 mismatches).
For WGBS, duplicate reads are marked and removed with Picard (Picard) (v2.20.2). This step is not performed for ERRBS.
Alignments below a MAPQ threshold are removed with samtools (Li et al., 2009) (v1.2) and the
parameters: -q 10.
MethylDackel (MethylDackel) (v0.4.0) then calls
methylation rates with parameters
-d 5 -D 2000 --mergeContext.
In the event of a BS/oxBS or BS/TAB library preparation, methylation mark deconvolution is then performed using the MLML2R R package (Qu et al., 2013). Briefly, methylated and unmethylated counts from bisulfite-only treated samples and oxidative-bisulfte treated samples are extracted and passed to MLML2R::MLML() to determine the levels of methylcytosine (mC), hydroxymethylcytosine (hmC), and cytosine (C) using the exact method provided in the package.
Differential Testing
Without Mark Deconvolution
Differential methylation testing is performed with the MethylSig R Bioconductor package which performs group-versus-group tests using a beta-binomial approach to calculate differential methylation statistics, accounting for variation among replicates within each group (Park et al., 2014). Alternately, differential methylation testing can be performed with the DSS R bioconductor package which is a beta-binomial approach with arcsine link function to test under general experimental design (Wu and Park, 2016).
With Mark Deconvolution
We use the gamlss R package to identify differentially methylated probes (DMPs) by fitting a beta-regression model on the beta-values (Stasinopoulos and Rigby, 2007). The DMPs are then annotated to CpG island and genic annotations using the annotatr R Bioconductor package (Cavalcante and Sartor, 2017).