EPIC Methylation Array
UM Bioinformatics Core
2023-08-29
Methods
QC
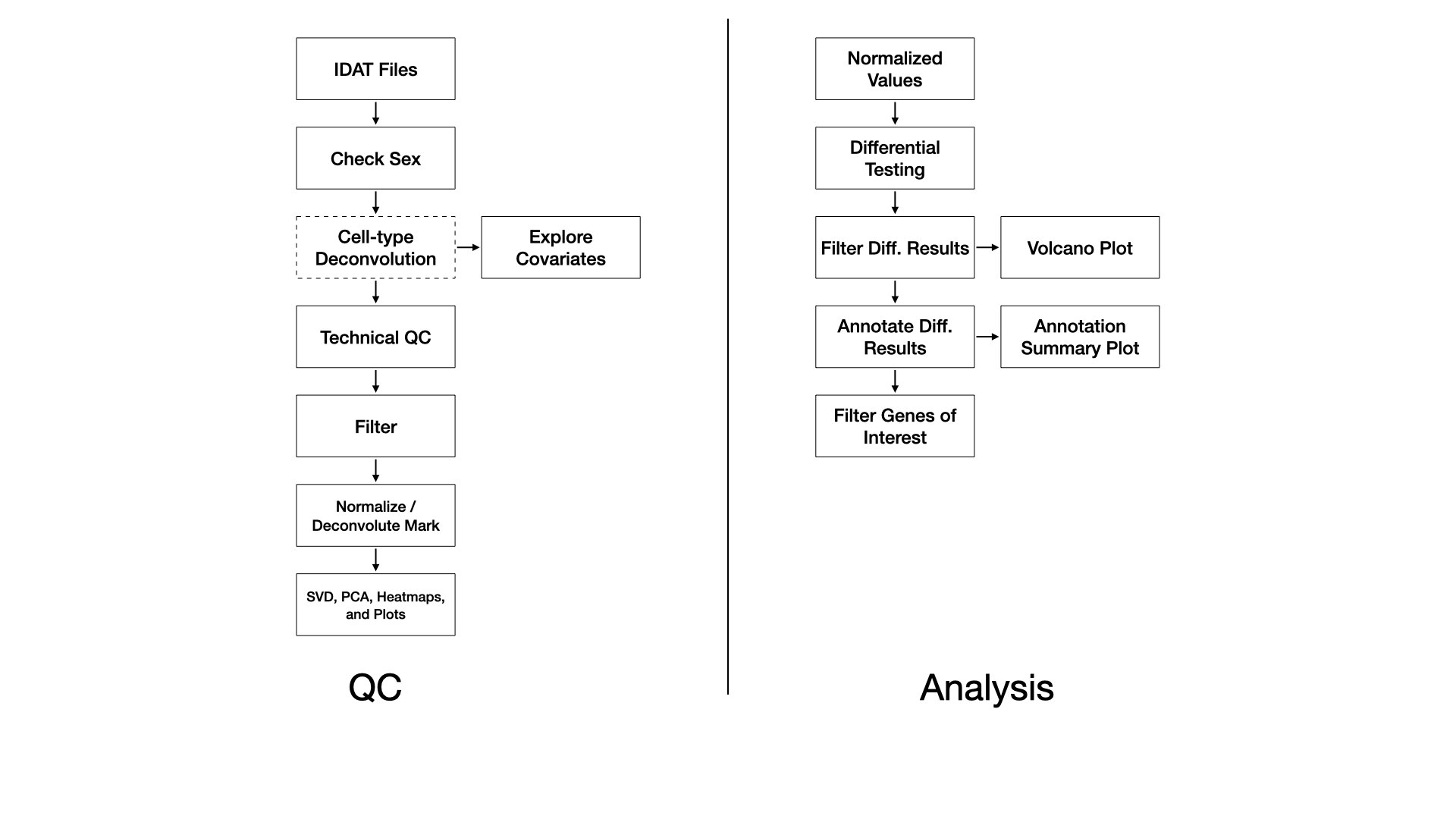
Samples are processed with the SeSAMe R Bioconductor package (Zhou et al., 2018). Briefly, red/green IDAT files are read and processed according to the preparation code. Experiment-independent masking occurs with the quality mask code (“Q”), and accounts for probes containing SNPs, with known cross-hybridization issues (Pidsley et al., 2013), and with other issues. Experiment-dependent is done according to the p-value with out-of-bound array hybridization (pOOBAH) algorithm (“P”), which is an improvement on detection p-value filtering (Zhou et al., 2018). Non-linear dye bias correction (“D”) is performed followed by background correction with the NOOB method (“B”) (Fortin et al., 2014). This processing pipeline constitutes a within-array normalization procedure. We note that recent studies have indicated within-array normalization with dye-bias correction and NOOB performs as well or better than between-array normalization procedures (Welsh et al., 2023).
A probe has with pOOBAH masking (p-value < 0.05) in more than 5% of samples it is removed. Similarly, if a sample has more than 10% of probes pOOBAH masked, then that sample is removed.
Cell type deconvolution of whole blood or cord blood may be performed with the FlowSorted.Blood.EPIC or FlowSorted.CordBlood.450k R Bioconductor package, respectively, using a modified version of the Houseman method (Houseman et al., 2012).
In the event of a BS/oxBS or BS/TAB library preparation, methylation mark deconvolution may be performed using the MLML2R R package (Qu et al., 2013). Briefly, methylated and unmethylated channel matrices from bisulfite-only treated samples and oxidative-bisulfte treated samples are extracted and passed to MLML2R::MLML() to determine the levels of methylcytosine (mC), hydroxymethylcytosine (hmC), and cytosine (C) using the exact method provided in the package.
Differential Testing
Without Mark Deconvolution
For each comparison we use the limma R Bioconductor package to identify differentially methylated probes (DMPs) by fitting a linear model on the M-values whose standard errors are then moderated using an empirical Bayes model (Ritchie et al., 2015). The DMPs are then annotated to CpG island and genic annotations using the annotatr R Bioconductor package (Cavalcante and Sartor, 2017).
With Mark Deconvolution
For each comparison we use the gamlss R package to identify differentially methylated probes (DMPs) by fitting a beta-regression model on the beta-values (Stasinopoulos and Rigby, 2007). The DMPs are then annotated to CpG island and genic annotations using the annotatr R Bioconductor package (Cavalcante and Sartor, 2017).